Density Functional Theory
Density Functional Theory (DFT) calculations have shaped material science with their ability to probe nanoscale properties and thus revealing core fundamental thermodynamic and kinetic material aspects. DFT offers an efficient and adequately accurate way to numerically approach the Schrödinger equation of nanoscale systems up to a few hundred atoms. Determining the electron density of the microstructure is the key to calculating macroscopic battery-related properties such as phase stability, voltage, and diffusion. In this context, we are active in DFT simulations, including ab-initio molecular dynamics, as an in-depth tool for understanding and improving battery performance. Our work revolves around computationally complementing and guiding experiments, for instance, characterization with XRD, NMR and, NR, and facilitating material predictions for electrochemical system optimization, for example, novel coatings for long-lasting interface stability in solid-state batteries. The software of expertise in our group is the commercial DFT packages VASP1 and CASTEP2.
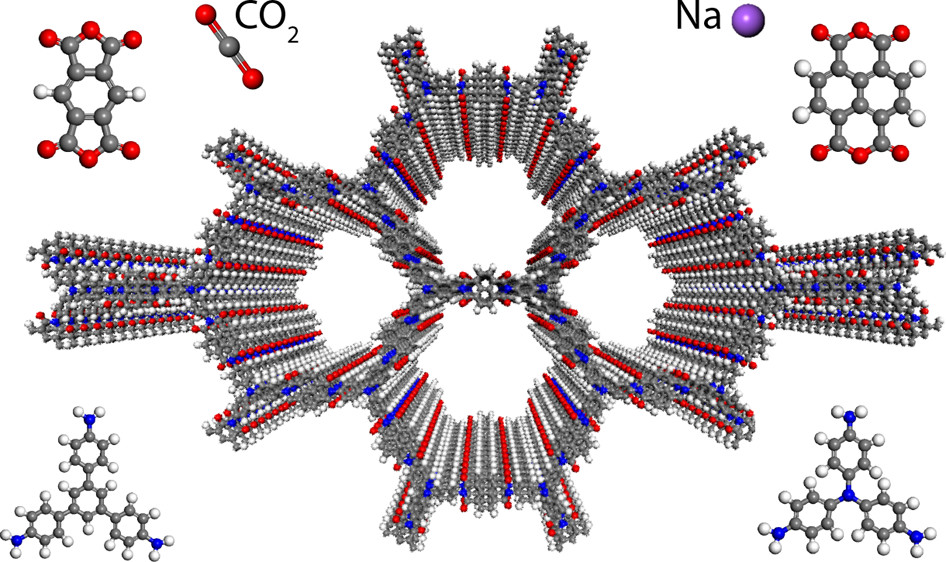
An example of detailed DFT structural characterization in covalent organic framework (COF) materials. It is revealed that the imide bonds prefer to form at an angle with one another, breaking the 2D symmetry, which shrinks the pore width elongates the pore walls.3
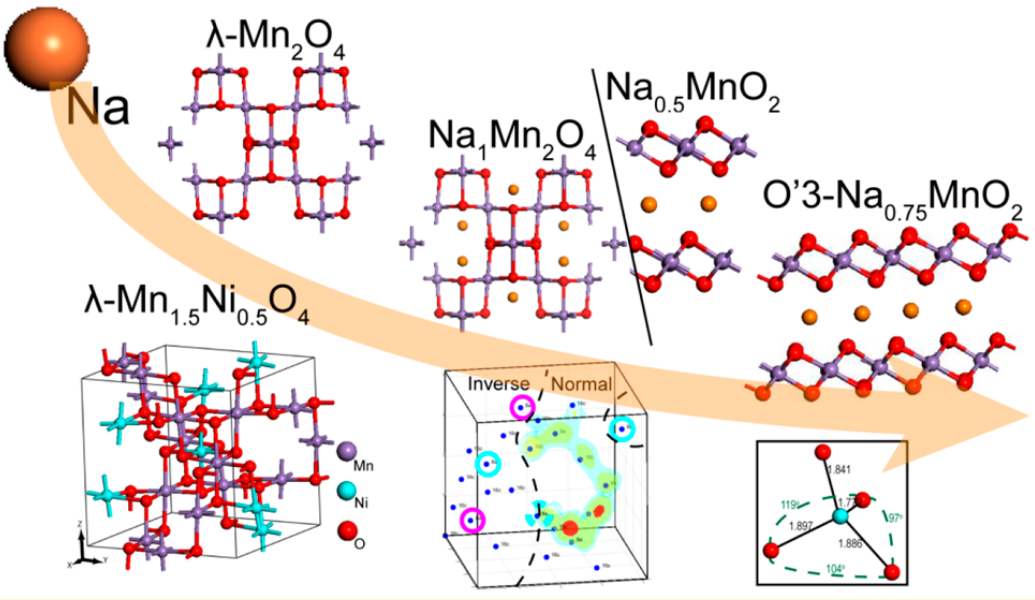
An example of DFT investigation on the thermodynamics of spinel and layered NaMnO2 structures. We followed the sodiation pathway of promising Na-ion electrode materials. Determination of the formation enthalpies revealed the metastable phases along sodiation matching the observed voltage trends.4
References
1. Computational Materials Science 1996 6, 15−50
2. Z. Kristallogr. 2005 220,567–570
3. Chemistry of Materials 2021 33, 818-833
4. Chemistry of Materials 2018 30, 6646-6659